The NER–STING Axis: When DNA Repair Becomes Immune Signaling A Perspective for the Turkish Journal of Immunology









When I began studying how cells repair ultraviolet (UV)-induced DNA damage, the objective was precise and mechanistic: to understand how damaged DNA is recognized and corrected at the molecular level. Through detailed biochemical analyses, nucleotide excision repair (NER) was defined as the pathway that removes bulky DNA lesions as short oligonucleotides of approximately 27–29 nucleotides in length. Subsequent genome-wide analyses of excision products revealed that these fragments can range from approximately 24–32 nucleotides, with the majority clustering around 26–27 nucleotides. This mechanistic understanding established NER as a central guardian of genomic stability (1).
For many years, the biological implications of NER were considered primarily within the framework of mutation prevention and cancer biology. The excised DNA fragments were viewed as transient intermediates–necessary products of lesion removal that were subsequently degraded after fulfilling their role in repair. The immune system was not part of this discussion.
However, as scientific fields evolve, so does the interpretation of earlier discoveries.
In recent years, increasing attention has been directed toward the relationship between DNA damage responses and innate immune signaling. The cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway, now recognized as a principal sensor of cytosolic DNA, activates TBK1, induces IRF3 phosphorylation, and drives type I interferon production (2). While initially characterized as a defense mechanism against viral DNA, this pathway also responds to endogenous DNA species under specific conditions (3).
Ultraviolet radiation provides a useful model for exploring this intersection. UV light induces cyclobutane pyrimidine dimers and 6–4 photoproducts in DNA. Through the coordinated action of damage recognition factors, helicases, and endonucleases, the damaged DNA segment is excised as an approximately 26–27 nt single-stranded DNA (ssDNA) oligonucleotide. Under normal circumstances, these excision products are efficiently processed. Yet cellular stress, altered regulatory pathways, or disruptions in downstream handling may allow these excised ssDNA fragments to access the cytoplasm, where they have the potential to engage innate immune sensors (Figure 1) (4).
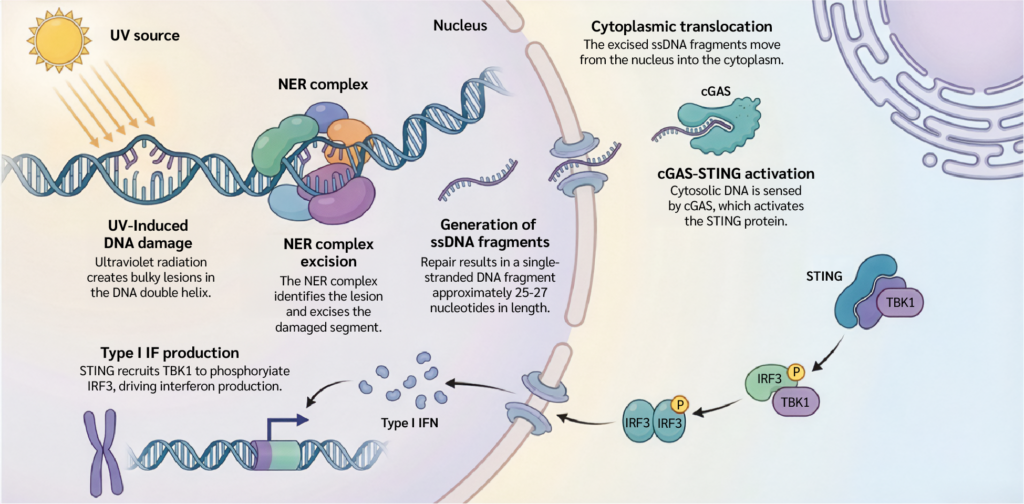
Figure 1.
The NER–STING axis: Connecting DNA repair to innate immunity.
The study by Kemp et al. (5) in 2015 offered important insight into this connection. Their findings demonstrated that UV irradiation potentiates STING-dependent signaling through deregulation of UNC-51–like kinase 1 (ULK1), a kinase associated with autophagy. This observation suggested that the cellular response to UV damage extends beyond DNA lesion repair and includes modulation of immune signaling pathways.
These findings do not alter the fundamental mechanics of NER. Rather, they expand its biological context. Genome stability and immune surveillance, long studied as distinct systems, may in fact represent integrated components of cellular defense. From an evolutionary perspective, this integration is logical. Cells must defend not only against pathogens but also against environmental and endogenous threats that compromise genomic integrity.
The implications of the NER–STING interface extend across multiple areas of immunology.
In sterile inflammation, endogenous DNA fragments generated during stress may contribute to interferon responses even in the absence of infection. In tissues chronically exposed to ultraviolet light, repair-associated DNA species could influence local immune signaling.
In autoimmunity, excessive type I interferon activity is a hallmark of several diseases. While apoptotic debris and mitochondrial DNA have been proposed as sources of immunostimulatory DNA, repair-derived ssDNA fragments may warrant further investigation as contributors to interferon amplification (6).
In cancer biology, DNA-damaging therapies enhance tumor immunogenicity not only by increasing mutation burden but also by activating cytosolic DNA sensing pathways. Accumulation of DNA fragments in the cytoplasm triggers the cGAS–STING axis, leading to type I interferon production and the promotion of anti-tumor immune responses (7). In this context, DNA repair–derived oligonucleotides, originally characterized through nucleotide excision repair studies (1), may represent an additional endogenous source of immunostimulatory DNA. Thus, DNA repair and innate immune activation emerge as interconnected components of the cellular stress response.
Aging represents another dimension in which these systems intersect. Accumulation of DNA damage over time may lead to persistent low-level activation of innate immune pathways, contributing to chronic inflammatory states. Understanding how repair intermediates are processed or contained may therefore have broader relevance for immune regulation across the lifespan.
These developments illustrate a broader principle in science: mechanistic discoveries often acquire additional biological meaning as new technologies and conceptual frameworks emerge. When nucleotide excision repair was first characterized, its significance lay in preventing mutagenesis and preserving genomic fidelity. Today, it occupies a place within a larger network that links DNA damage responses to innate immune signaling.
The cell does not divide its defense systems into the categories that we assign in academic disciplines. Genome maintenance and immune vigilance are coordinated elements of a unified protective strategy. The emerging NER–STING axis underscores this integration and invites continued interdisciplinary investigation.
The story of nucleotide excision repair began with ultraviolet light and damaged DNA. It now extends into the biology of innate immunity. This expansion does not redefine the pathway; rather, it reveals its broader relevance within cellular defense architecture. As our understanding deepens, the connection between genome integrity and immune regulation will likely become even more refined.
Fundamental research, pursued with precision and persistence, can illuminate relationships that were not initially anticipated. The evolving dialogue between DNA repair and immunology represents one such example.
Future studies should clarify how nucleotide excision repair–derived ssDNA fragments are processed and whether they can access the cytoplasm under specific cellular conditions. Understanding how these repair intermediates intersect with innate immune sensing pathways may reveal new links between genome maintenance, inflammation, and disease. Elucidating this interface could open new perspectives in cancer immunology and immune regulation.
Ultimately, the emerging NER–STING axis reminds us that mechanisms originally evolved to preserve genome integrity may also serve as unexpected bridges between DNA repair and innate immune defense.
References
Sancar A. Excision repair in mammalian cells. J Biol Chem. 1995;270(27):15915–8. [CrossRef]
Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–9. [CrossRef]
Ablasser A, Chen ZJ. cGAS in action: Expanding roles in immunity and inflammation. Science. 2019;363(6431):eaat8657. [CrossRef]
Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular nucleic acid detection in autoimmunity. Nat Rev Immunol. 2017;17(4):264–75. [CrossRef]
Kemp MG, Lindsey-Boltz LA, Sancar A. UV light potentiates STING-dependent innate immune signaling through deregulation of ULK1 (Unc51-like kinase 1). J Biol Chem. 2015;290(19):12184–94. [CrossRef]
Harding SM, Benci JL, Irianto J, et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–70. [CrossRef]
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–52. [CrossRef]
VOLUME
,
ISSUE
Correspondence
Published
Suggested Citation
DOI
License
