Non-Infectious Gastrointestinal Tract Complications in Patients with Common Variable Immunodeficiency Disorders: Clinical Perspectives, Immunologic Insights and Therapeutic Approaches










Abstract
Keywords:
Common, variable, immune, deficiency CVID enteropathy, in, CVID Gastrointestinal, tract, inflammation, in, CVIDHighlights
- Common variable immunodeficiency (CVID) is an inborn error of immunity with both infectious and non-infectious complications that significantly impact morbidity and mortality.
- Gastrointestinal involvement is common in CVID and presents with a wide spectrum of non-infectious pathologies, often resembling inflammatory bowel disease but with unique histopathological features.
- Common variable immunodeficiency enteropathy is marked by T helper 1 (Th1)-driven inflammation, impaired T cell responsiveness, and decreased natural killer (NK) cell levels, which together may contribute to chronic gastrointestinal symptoms.
- There is currently no consensus on the definition or management of CVID enteropathy, and no established treatment guidelines exist.
- Personalized, multidisciplinary, and stepwise treatment strategies based on symptom severity, histopathology, comorbidities, and patient preferences are recommended.
Introduction
Inborn errors of immunity (IEI), formerly known as primary immunodeficiency (PID), represent a diverse group of disorders, marked by varying levels of immunodeficiency and immune dysregulation (1). Although the prevalence varies by specific disease, the overall estimated prevalence is approximately 1/1200, likely lower than the true rate due to underdiagnosis (2,3). Among IEIs, predominantly antibody deficiencies (PADs), especially selective IgA deficiency and common variable immunodeficiency disease (CVID), are the most common (2-5).
Inborn errors of immunity are associated with increased risk of a wide range of comorbidities and complications, broadly classified as either infectious or non-infectious (6). Depending on the underlying disease and the affected component of the immune system, any type of infectious complication, including bacterial, viral, fungal, or opportunistic infections, may occur; these infections tend to recur and can be severe (7). Noninfectious complications comprise autoimmunity, splenomegaly, chronic lung diseases such as asthma or bronchiectasis, enteropathy, granulocytic and lymphocytic organ infiltrations, granulomas, and malignancy (8). These complications result in long-term sequelae, including structural damage due to infections such as bronchiectasis and progressive organ dysfunctions driven by immune dysregulation (9,10). These complications and their consequences are closely related to both mortality and a significant burden of the disease on patients, their families, as well as health care systems (2,4).
Mortality rates in IEIs vary based on the specific underlying diseases and the associated complications (2). The age-adjusted mortality rate was estimated to be 0.43/1,000,000 people in an epidemiological study from the United States, identifying antibody deficiency as the most common cause of death. The leading contributing factors were infections (34.6%), followed by respiratory and cardiovascular complications (17.4% and 11.4%, respectively) (11). Another epidemiological study from Europe indicated that annual and premature mortality rates in patients with CVID were 1.7–3 times higher than in the general population. Chronic respiratory disease and malignancy were the leading causes of death in this cohort (5). Notably, respiratory complications followed by gastrointestinal complications are the most common causes of morbidity in CVID patients (12).
Given this impact, it is crucial to thoroughly understand the complications from all perspectives, including clinical features, underlying mechanisms, and treatment. In this review, we examine the various aspects of gastrointestinal (GI) tract manifestations in adult patients with CVID, with a focus on non-infectious complications.
Common Variable Immunodeficiency
Common variable immunodeficiency is the most commonly occurring symptomatic IEI, with a prevalence ranging from 1/10,000 to 1/50,000 (13,14). It is a complex disorder characterized by heterogeneous clinical features and impaired antibody production due to a B-cell defect. Although the underlying pathomechanism is not well understood, in addition to the monogenic defects identified in approximately 30% of patients, T cell abnormalities related to non-infectious complications may accompany B cell dysfunction (15). In the latest update to the classification of IEIs, PADs were categorized into four main groups: 1) Severe reduction in all immunoglobulin (Ig) isotypes with profoundly decreased or absent B cells; 2) Severe reduction in at least two Ig isotypes with normal or low B cells; 3) Severe reduction in IgG and IgA with normal/elevated IgM and normal counts of B cells; 4) Isotype, light chain, or functional deficiencies with generally normal counts of B cells (16). Common variable immunodeficiency is further categorized into two main groups: 1) Common variable immunodeficiency with no gene defect specified and 2) Specific disorders caused by monogenic defects such as activated phosphoinositide-3 kinase delta syndrome (APDS), PTEN, CD19, CD81, CD20, CD21, TACI, BAFFR, TWEAK, TRNT1, ICAROS or NFKB1 and NFKB2 deficiencies (16).
Clinical Features
The most common initial clinical manifestation of CVID is recurrent sinopulmonary infections mainly caused by encapsulated bacteria. However, a substantial proportion of patients also suffer from diverse immune-dysregulation-related diseases, such as autoimmune or inflammatory disorders, allergies, granulomatous or lymphocytic inflammation of various organs, benign lymphoproliferation, and malignancy, with multiple organ systems potentially affected (1,2). On the other hand, in some patients, various non-infectious manifestations such as enteropathy, granulomatous diseases, malignancy, or autoimmune diseases may precede infectious symptoms as the initial clinical presentation (9).
Diagnosis
Common variable immunodeficiency encompasses a heterogeneous group of predominantly antibody deficiency syndromes and is generally considered a diagnosis of exclusion. Unfortunately, no clinical features or laboratory tests are pathognomonic for CVID. Therefore, clinical diagnosis and diagnostic criteria become essential for identifying cases. Several expert groups proposed distinct diagnostic criteria for CVID (8,17,18). In the late of 90s, CVID was defined by experts as the presence of hypogammaglobulinemia (a marked decrease in IgG, at least two standard deviations below the normal range for age), a marked decrease in IgM or IgA, impaired vaccine response or absence of isohemagglutinins, and the exclusion of defined causes of hypogammaglobulinemia in patients whose immunodeficiency began after 2 years of age (17). However, some authors did not agree with the strict necessity of a poor vaccine response for the diagnosis. In 2019, the European Society for Immunodeficiencies (ESID) reported clinical diagnostic criteria for a probable diagnosis of CVID (=clinical diagnosis) (Table 1). In this expert consensus, poor vaccine response is included among the supportive laboratory findings (18).

Previously, genetic analysis was recommended for patients with complications such as autoimmunity or malignancy to investigate underlying genetic defects in CVID, and was not usually performed in patients with only infections (8). With advances in genetic testing and a deeper understanding of underlying mechanisms, it has been demonstrated that some monogenic defects may respond to specific treatments, including enzyme replacement, targeted monoclonal antibodies, or stem cell transplantation (18). Therefore, genetic testing should be performed in all patients.
Diagnostic delay is a significant challenge in the management of CVID patients. A multicenter study involving 2212 patients from various parts of Europe reported a delay in diagnosis of five to seven years (6). From Türkiye, a case series of 44 pediatric patients reported a mean diagnostic delay of 4.6 years (19). On the other hand, another study from Türkiye reported an average delay in diagnosis of 14.9 years, highlighting the significance of this issue in adults (20). Diagnostic delay is crucial, as it may lead to a delay in specific treatment and the development of complications.
Treatment
In CVID, the primary cornerstone of the treatment is Ig replacement therapy (IgRT), which is highly effective in preventing infectious diseases and, consequently, reducing infection-related mortality. However, IgRT has a limited impact on the non-infectious complications (21). Furthermore, the evidence of the effectiveness of immunosuppressive or immunomodulatory medications in treating inflammatory and autoimmune complications is inadequate. A better understanding of the underlying molecular mechanisms of the complications can pave the way for specific, safe, and effective treatment modalities. Therefore, comprehensive studies about complications are essential.
Gastrointestinal Tract Involvement in CVID
Gastrointestinal tract complications leading to malabsorption are a significant clinical feature in CVID. The gastrointestinal tract is the largest organ-system tract, playing a crucial role in the body’s immune system. Therefore, GI involvement is not unusual. The frequency of GI tract complications ranges between 9% to 34% (6,22-24). Gastrointestinal tract involvement can be categorized into two main groups: infectious and non-infectious complications. Since the aim of our review is to highlight and describe non-infectious complications, infectious complications will not be discussed in this context.
Non-infectious Complications
In CVID, non-infectious pathologies are encountered across a wide spectrum of varying severity, affecting any part of the GI tract (25). Gastrointestinal tract complications and malabsorption are related to more than two-fold increased mortality in CVID patients (22).
Chronic or intermittent diarrhea is the most common GI tract-associated symptom (25). Bloating and pain are also frequently encountered (26). The clinical manifestations may mimic inflammatory bowel diseases (IBD), including both ulcerative colitis and Crohn’s disease, Whipple’s disease, collagenous colitis, and celiac disease (27-31). IBD-like manifestations, commonly after CVID diagnosis, can be seen in up to 10% of patients (30, 32). However, some patients presented with GI manifestations can be initially misdiagnosed with IBD since it can be challenging to differentiate the inflammatory features of IBD and CVID (27,31,33). Furthermore, duodenal histopathology can resemble celiac disease in terms of increased intraepithelial lymphocytosis with or without villous atrophy. But gluten withdrawal does not work in CVID (25). Inflammation in CVID enteropathy is thought to be multifactorial, and various factors, including immunodeficiency, immune dysregulation, autoimmunity, GI microbiota, genetics, cancer, and infections, may contribute to this inflammation (34). Interestingly, some patients may exhibit gastrointestinal inflammation in the absence of symptoms, whereas others may present with symptoms despite lacking histological evidence of inflammation (26,27).
The definition of GI tract disease in CVID, also known as CVID enteropathy, has not yet reached a consensus. Some authors combine clinical manifestations with histopathological findings, while others also include GI infections (29,35-41). The features range from mild to moderate, characterized by chronic diarrhea or increased intraepithelial lymphocytosis, and from no malnutrition or weight loss to severe cases with weight loss, malnutrition, extensive IgG loss, and increased intraepithelial lymphocytosis or histopathology, such as GVHD (25). Since the management is different, excluding infectious diseases and including both clinical and histopathological features in the definition seems logical.
The discrepancy in the definition of CVID enteropathy can lead to problems in both research studies and treatment. Therefore, precise and detailed characterization of disease phenotypes in clinical reports may lead to a better understanding and help in standardization in future studies.
In CVID patients with GI involvement, the inflammation is very heterogeneous and can be observed in any part of the GI tract, leading to various diseases (Table 2). In the majority of patients with CVID, plasma cells are decreased or absent in the lamina propria regardless of the location of the inflammation in the bowel (33,41). Another common manifestation is nodular lymphoid hyperplasia (NLH), which is defined as the formation of nodules at least 1 mm in diameter with a germinal center. NLH can be frequently detected in both the small and large intestine of CVID patients, whereas it is rarely observed in immunocompetent adults and usually occurs secondary to infections (29). The etiopathology of NLH in CVID is not clearly known. Although it is usually associated with the presence of abdominal symptoms, it is sometimes detected in asymptomatic patients coincidentally (33,41,42). Therefore, rather than representing a primary pathologic manifestation, it may be related to immune dysregulation.
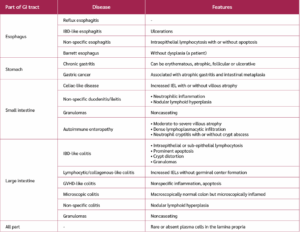
Esophageal involvement is rare in patients with CVID, and its exact prevalence remains unclear. The most frequently reported manifestation is Candida esophagitis. Non-infectious esophageal complications include reflux esophagitis, intraepithelial lymphocytosis, esophageal ulcerations, and Barrett's esophagus (33,43).
The frequency of chronic gastritis ranges from 3% to 27% in various cohorts (25,26,35,41). Helicobacter pylori, EBV, or CMV infections were infrequently associated with chronic gastritis in CVID patients. This remarkable information suggests that the impact of continuous gastric mucosal inflammation is most likely due to immune dysregulation rather than infections in CVID (25). Gastric cancer, closely related to atrophic gastritis and intestinal metaplasia, represents another pathological condition of the stomach and has a significant impact on mortality (9,44).
The most common histopathological finding in the duodenum is increased intraepithelial lymphocytosis with or without villous atrophy, which is often less prominent than in celiac disease (33). The major distinguishing feature is the absence of plasma cell infiltration in CVID (33). Furthermore, neutrophil infiltration and follicular lymphoid hyperplasia can be observed in CVID, whereas they are rare in celiac disease (33,41,45). Patients with CVID are also insensitive to a gluten-free diet (41). On the other hand, to explore the relation between celiac disease and celiac-like findings in CVID, some authors studied the celiac-associated HLA profiles, including HLA-DQ2 and HLA-DQ8, in CVID patients. In a study, these HLA profiles were detected in 20% of the patients, whereas in another study, no association was found, suggesting that celiac-like and true celiac disease are distinct entities (26,46).
In patients with CVID, colonic histopathological findings are diverse and can mimic several gastrointestinal disorders (40). These include patterns resembling inflammatory bowel disease (IBD), as well as lymphocytic or collagenous colitis, and GVHD-like colitis (40). Notably, inflammation may not be apparent macroscopically and can only be detected through microscopic examination, sometimes presenting as non-specific patterns. Classic IBD can be distinguished from IBD-like disease in CVID by the presence of plasma cells in the lamina propria (40).
Autoimmune neutropenia (AIN) is a rare but recognized hematologic complication in CVID and is frequently accompanied by other cytopenia such as immune thrombocytopenia or autoimmune hemolytic anemia (47). Patients with AIN appear to carry an elevated risk of enteric inflammation, including chronic enteropathy, crypt distortion, villous blunting, lymphoid aggregates, and opportunistic gut infections such as cytomegalovirus colitis, especially in the context of neutropenia-related mucosal vulnerability (47). According to the United States Immunodeficiency Network (USIDNET) registry study, patients with AIN exhibited a 2.1 fold and 3.4 fold increased risk of developing enteropathy and autoimmune enteropathy, respectively. In this study, the term "enteropathy" encompassed a broad spectrum of gastrointestinal inflammatory conditions, including gastroenteropathy, enterocolitis, lymphocytic colitis, chronic and atrophic gastritis, gastroenteritis, enteritis, and duodenitis, closely resembling the histopathological features of broader CVID-associated enteropathy and IBD-like lesions (48).
Although some cases of eosinophilic inflammation in the gut of patients with CVID have been reported, the role of eosinophilic infiltration as part of the inflammatory process in GI involvement in CVID remains poorly defined (29,41,45).
Immunologic Features
Although the underlying immunopathogenesis in CVID enteropathy is not clearly defined, it is believed that a combination of defective humoral immunity, impaired regulatory T cell function, altered microbiota, and abnormal cytokine responses plays a significant role. Studies have shown that CVID patients with symptomatic GI tract inflammation have distinctive immunologic abnormalities (34,38). In symptomatic patients with CVID and GI inflammation, the level of natural killer (NK) cells in peripheral blood was lower than that of CVID patients without GI-related symptoms. Additionally, it has been demonstrated that a skewing towards a pro-inflammatory Th1 response, accompanied by elevated levels of cytokines such as interferon-γ and IL-12, contributes to chronic mucosal inflammation (34). Th1 cytokine overproduction is closely linked to GI symptoms, particularly with malabsorption (34). This Th1 response was distinct from the Th1 profile seen in Crohn's disease, where IL-23 and IL-17 responses were also involved (34). Also, while T cell hyperresponsiveness to intestinal flora antigens in the gastrointestinal tract is a hallmark of Crohn’s disease, T cell hyporesponsiveness is characteristic of CVID (34). These findings support the notion that they are different entities.
A successful personalized, targeted therapeutic approach should be guided by the underlying immunopathogenesis. Although a prominent Th1 response is observed in both diseases, the cytokine profiles differ. Anti-IL-23 therapies (targeting the p19 subunit) may not be effective in inflammatory settings where IL-12 plays a dominant role (34). Accordingly, further in-depth and comprehensive studies are warranted to fully clarify these mechanisms.
Treatment of Non-infectious Complications
Currently, no established guidelines exist for managing the non-infectious complications of CVID. Furthermore, patients were often unresponsive to standard therapeutic agents used in inflammatory conditions of the bowel. Usually, IgRT is insufficient for either preventing or treating non-infectious GI manifestations (40). According to a meta-analysis, 70% of patients required an additional agent for the treatment of GI symptoms, and only a few cases were responsive to an increased dose of Ig without additional medications (49). Steroids, immunosuppressive agents, and biological agents have been used, and inconsistent results regarding the effectiveness of these treatments have been published to date (50,51).
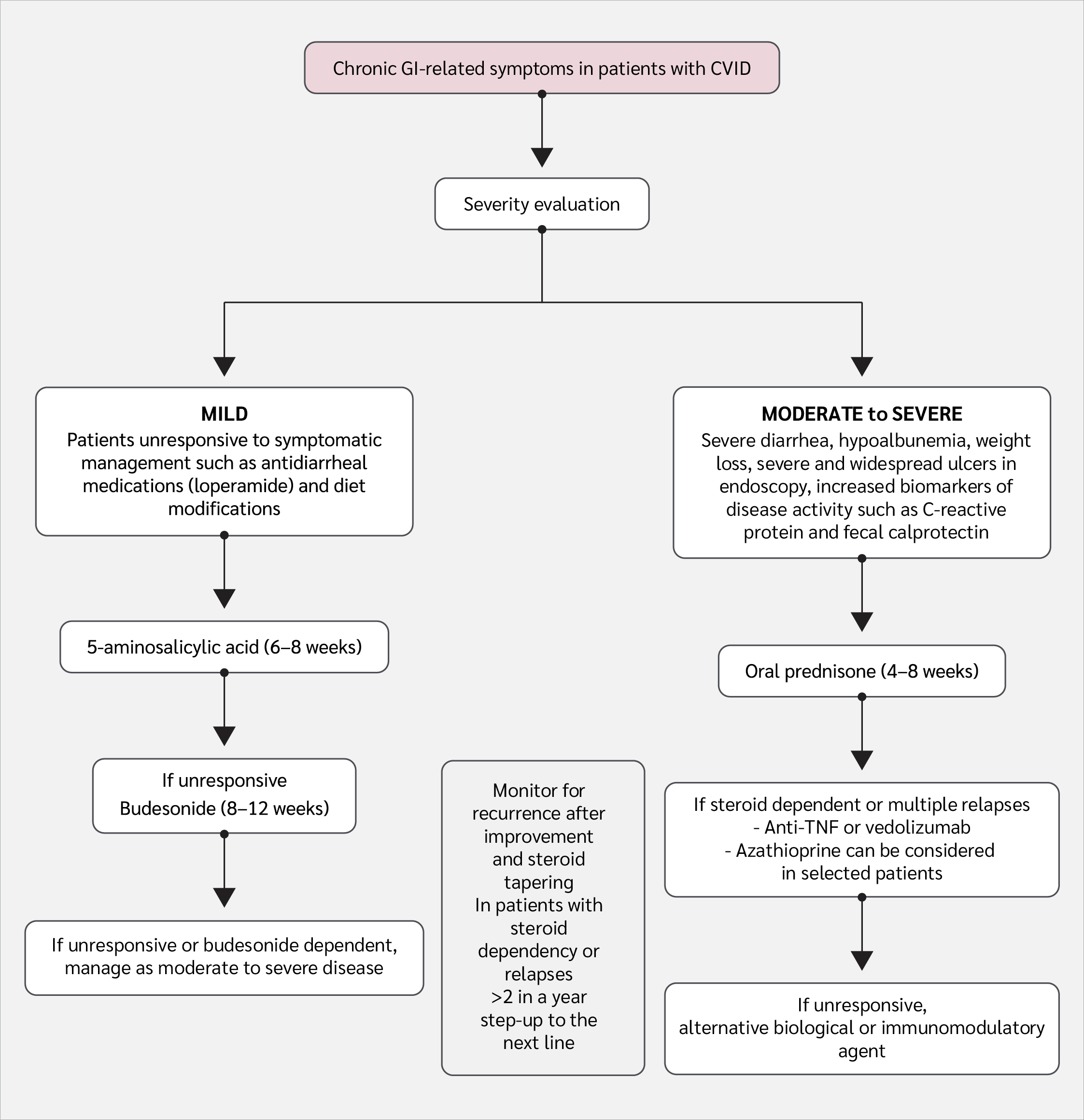
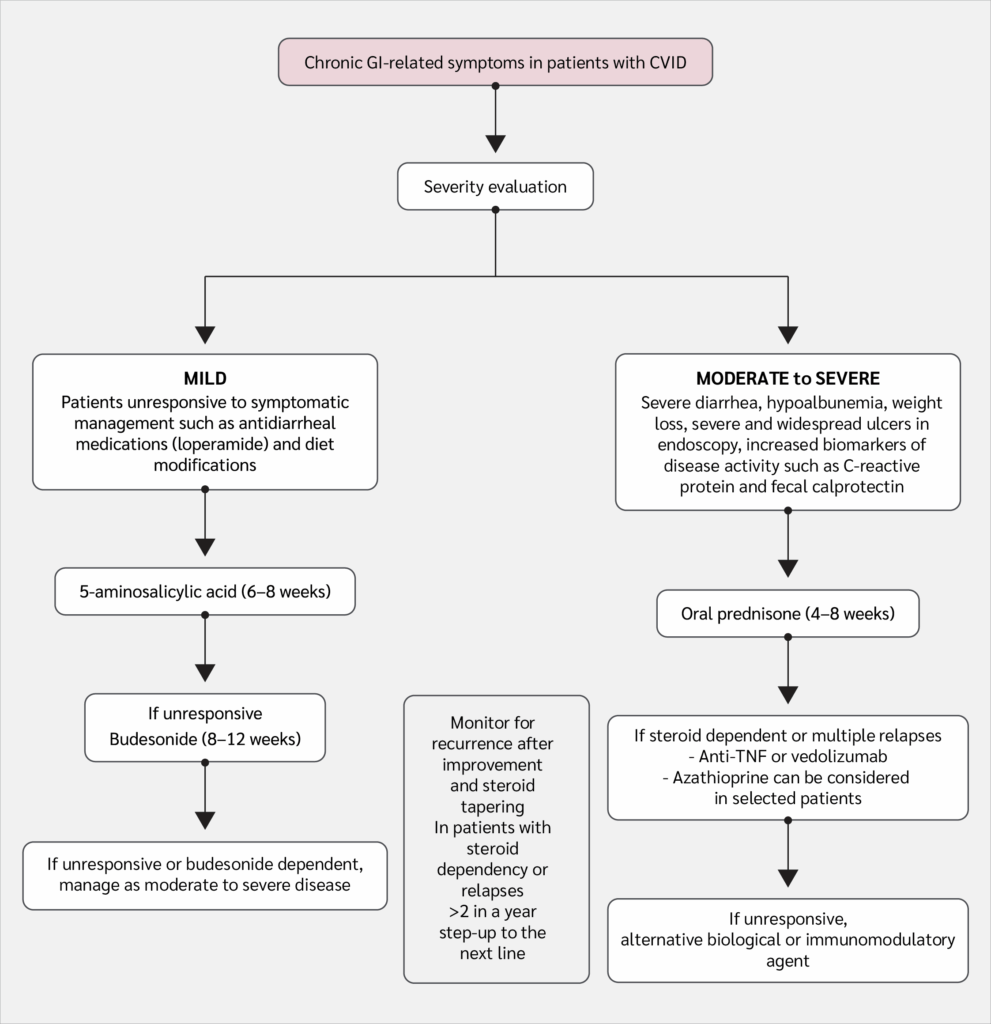
Figure 1:
Non-infectious gastrointestinal tract diseases in common variable immunodeficiency (CVID).
Personalized treatment with a multidisciplinary and stepwise approach, based on the severity of symptoms, histopathologic findings, comorbidities, and patient preferences, is proposed in an expert opinion approach (Figure 1). Moderate to severe disease is defined as the presence of severe diarrhea leading to hypoalbuminemia, weight loss, extensive and severe ulcers in endoscopic evaluation, and increased biomarkers of disease activity such as CRP and fecal calprotectin (50). Accordingly, the absence of these clinical features in symptomatic patients can be considered indicative of mild disease (50). Authors of this manuscript recommend oral prednisone for 4–8 weeks as first-line therapy for moderately or severely affected patients. If the disease becomes steroid-dependent or multiple relapses occur, vedolizumab or infliximab is recommended (50). If there is no response to these agents, alternative biologic or immunomodulatory therapies are suggested as second- and third-line options, respectively. In mildly affected patients, 5-aminosalicylic acid (for 6–8 weeks) is recommended as the first-line therapy (50). If patients are unresponsive, treatment should be stepped up to oral budesonide (for 8–12 weeks); if this also fails, patients should be managed as moderately or severely affected. The use of biologics before thiopurines is recommended, as inconsistent results have been reported with thiopurines, whereas anti-TNF and non-anti-TNF biologic agents have shown more consistent success (50). In line with these suggestions, a case series showed that biologics had the highest success rate in achieving complete remission (52). Azathioprine was used in three patients, and complete remission was achieved in one of them (52). Although this approach appears logical based on the results, given the high economic costs of the biologics, thiopurines may be considered as an alternative to biologics in selected patients.
Other Potential Treatment Modalities
For patients with CVID-associated enteropathy who do not respond adequately to the mentioned therapies, alternative and emerging treatment strategies may offer additional clinical benefit. Pediatric and adult CVID patients with refractory diarrhea were treated with oral Ig treatment (53). Furthermore, in a limited number of IEI patients with AIN-related enteropathy, cyclosporine, tacrolimus, cyclophosphamide, mycophenolate mofetil, abatacept, and hematopoietic stem cell transplantation were used, indicating that these modalities can be promising options in CVID patients with refractory autoimmune enteropathy (54).
Mesenchymal stem cell infusion has emerged as a potential therapeutic option in autoimmune enteropathy. A recent case in an adult with autoimmune enteropathy demonstrated short-term remission following this treatment (55). However, there are no further comprehensive studies that confirm the effectiveness of these treatment options.
Conclusion
Non-infectious complications affecting any organs or tissues and associated with increased morbidity and mortality in CVID patients require heightened clinical attention, careful evaluation, and prompt management through a multidisciplinary, tailored therapeutic approach. Unfortunately, many aspects of the GI tract involvement in CVID mimicking other GI diseases, including celiac disease or IBD, and leading to a two-fold increased mortality rate, are not well known. This review article highlights the urgent need for studies that will help elucidate the underlying pathophysiology, thereby clarifying and facilitating the management of affected patients.
Ethical Approval
N.A.
Informed Consent
N.A.
Peer-review
Externally peer-reviewed
Author Contributions
Concept – S.D., F.S.B., O.O.Y., N.K., D.Ü., A.G., G.D.; Design – S.D., F.S.B., O.O.Y., N.K., D.Ü., A.G., G.D.; Supervision – S.D., G.D.; Data Collection and/or Processing – S.D., F.S.B., O.O.Y., N.K., D.Ü., A.G., G.D.; Analysis and/or Interpretation – S.D., F.S.B., O.O.Y., N.K., D.Ü., A.G., G.D.; Literature Review – S.D., O.O.Y., D.Ü., G.D.; Writer – S.D., F.S.B., O.O.Y., N.K., D.Ü., A.G., G.D.; Critical Reviews – S.D., F.S.B., O.O.Y., N.K., D.Ü., A.G., G.D.
Conflict of Interest
The authors declare no conflict of interest.
Financial Disclosure
The authors declared that this study has received no financial support.
References
Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473-507. [CrossRef]
Hsueh JC, Van Hersh AT, Zhao W. Immunodeficiency: burden of illness. Allergy Asthma Proc. 2024;45(5):294-8. [CrossRef]
Modell V, Quinn J, Orange J, Notarangelo LD, Modell F. Primary immunodeficiencies worldwide: an updated overview from the Jeffrey Modell Centers Global Network. Immunol Res. 2016;64(3):736-53. [CrossRef]
Abolhassani H, Azizi G, Sharifi L, Yazdani R, Mohsenzadegan M, Delavari S, et al. Global systematic review of primary immunodeficiency registries. Expert Rev Clin Immunol. 2020;16(7):717-32. [CrossRef]
Odnoletkova I, Kindle G, Quinti I, Grimbacher B, Knerr V, Gathmann B, et al; Plasma Protein Therapeutics Association (PPTA) Taskforce. The burden of common variable immunodeficiency disorders: a retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J Rare Dis. 2018;13(1):201. [CrossRef]
Gathmann B, Mahlaoui N; CEREDIH; Gérard L, Oksenhendler E, Warnatz K, Schulze I, et al; European Society for Immunodeficiencies Registry Working Party. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(1):116-26. [CrossRef]
GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385(9963):117-71. [CrossRef]
Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): Common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(1):38-59. [CrossRef]
Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al; Italian Primary Immunodeficiency Network. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27(3):308-16. [CrossRef]
Durandy A, Kracker S, Fischer A. Primary antibody deficiencies. Nat Rev Immunol. 2013;13(7):519-33. [CrossRef]
Fernández Pérez ER, Hunter M, Katial RK. United States trends in mortality rates for primary immunodeficiency diseases. J Allergy Clin Immunol Pract. 2019;7(3):1045-8. [CrossRef]
Velthof L, Geldof J, Truyens M, Van Dorpe J, Ferdinande L, De Vriendt C, et al. Gastrointestinal disease in common variable immunodeficiency disorder (CVID): Histological patterns, diagnostic clues and pitfalls for the pathologist and gastroenterologist. J Clin Med. 2025;14(2):497. [CrossRef]
Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, et al. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol. 2014;5:415. [CrossRef]
Çalişkaner AZ, Reisli İ, Arslan Ş, Uçar R, Ataseven H, Selçuk NY. Common variable immunodeficiency in adults requires reserved protocols for long-term follow-up. Turk J Med Sci. 2016;46(2):430-6. [CrossRef]
Abolhassani H, Hammarström L, Cunningham-Rundles C. Current genetic landscape in common variable immune deficiency. Blood. 2020;135(9):656-67. [CrossRef]
Poli MC, Aksentijevich I, Bousfiha AA, Cunningham-Rundles C, Hambleton S, Klein C, et al. Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. J Hum Immunol. 2025;1(1):e20250003. [CrossRef]
Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93(3):190-7. [CrossRef]
Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al; ESID Registry Working Party and collaborators. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract. 2019;7(6):1763-70. [CrossRef]
Nepesov S, Aygun FD, Firtina S, Cokugras H, Camcioglu Y. Clinical and immunological features of 44 common variable immunodeficiency patients: the experience of a single center in Turkey. Allergol Immunopathol (Madr). 2020;48(6):675-85. [CrossRef]
Özdemir E. Retrospective evaluation of adults with primary immunodeficiency disease. Postepy Dermatol Alergol. 2022;39(5):976-9. [CrossRef]
Quinti I, Soresina A, Guerra A, Rondelli R, Spadaro G, Agostini C, et al; IPINet Investigators. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol. 2011;31(3):315-22. [CrossRef]
Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650-7. [CrossRef]
Oksenhendler E, Gérard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al; DEFI Study Group. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46(10):1547-54. [CrossRef]
Cabañero-Navalon MD, Garcia-Bustos V, Nuñez-Beltran M, Císcar Fernández P, Mateu L, Solanich X, Carrillo-Linares JL, et al. Current clinical spectrum of common variable immunodeficiency in Spain: The multicentric nationwide GTEM-SEMI-CVID registry. Front Immunol. 2022;13:1033666. [CrossRef]
Andersen IM, Jørgensen SF. Gut inflammation in CVID: causes and consequences. Expert Rev Clin Immunol. 2022;18(1):31-45. [CrossRef]
Jørgensen SF, Reims HM, Frydenlund D, Holm K, Paulsen V, Michelsen AE, et al. A cross-sectional study of the prevalence of gastrointestinal symptoms and pathology in patients with common variable immunodeficiency. Am J Gastroenterol. 2016;111(10):1467-75. [CrossRef]
Malesza IJ, Malesza M, Krela-Kaźmierczak I, Zielińska A, Souto EB, Dobrowolska A, et al. Primary humoral immune deficiencies: Overlooked mimickers of chronic immune-mediated gastrointestinal diseases in adults. Int J Mol Sci. 2020;21(15):5223. [CrossRef]
Ho HE, Cunningham-Rundles C. Non-infectious complications of common variable immunodeficiency: Updated clinical spectrum, sequelae, and insights to pathogenesis. Front Immunol. 2020;11:149. [CrossRef]
van Schewick CM, Lowe DM, Burns SO, Workman S, Symes A, Guzman D, et al. Bowel histology of CVID patients reveals distinct patterns of mucosal inflammation. J Clin Immunol. 2022;42(1):46-59. [CrossRef]
Yong PF, Li H, Chung-Faye G, Ibrahim MA. Collagenous colitis in a patient with common variable immunodeficiency. J Investig Allergol Clin Immunol. 2008;18(6):482-3.
Albshesh A, Eder P, Ribaldone DG, Oldenburg B, de Boer NK, Mantzaris GJ, et al. Primary hypogammaglobulinaemia with inflammatory bowel disease-like features: An ECCO CONFER multicentre case series. J Crohns Colitis. 2022;16(1):91-7. [CrossRef]
Dion J, Malphettes M, Bénéjat L, Mégraud F, Wargnier A, Boutboul D,et al; DEFI study group. Campylobacter infection in adult patients with primary antibody deficiency. J Allergy Clin Immunol Pract. 2019;7(3):1038-41.e4. [CrossRef]
Daniels JA, Lederman HM, Maitra A, Montgomery EA. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): a clinicopathologic study and review. Am J Surg Pathol. 2007;31(12):1800-12. [CrossRef]
Mannon PJ, Fuss IJ, Dill S, Friend J, Groden C, Hornung R, et al. Excess IL-12 but not IL-23 accompanies the inflammatory bowel disease associated with common variable immunodeficiency. Gastroenterology. 2006;131(3):748-56. [CrossRef]
Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277-86. [CrossRef]
Li J, Jørgensen SF, Maggadottir SM, Bakay M, Warnatz K, Glessner J, et al. Association of CLEC16A with human common variable immunodeficiency disorder and role in murine B cells. Nat Commun. 2015;6:6804.[CrossRef]
Farmer JR, Ong M-S, Barmettler S, Yonker LM, Fuleihan R, Sullivan KE, et al; USIDNET Consortium. Common variable immunodeficiency non-infectious disease endotypes redefined using unbiased network clustering in large electronic datasets. Front Immunol. 2018;8:1740. [CrossRef]
Shulzhenko N, Dong X, Vyshenska D, Greer RL, Gurung M, Vasquez-Perez S, et al. CVID enteropathy is characterized by exceeding low mucosal IgA levels and interferon-driven inflammation possibly related to the presence of a pathobiont. Clin Immunol. 2018;197:139-53. [CrossRef]
van Schewick CM, Nöltner C, Abel S, Burns SO, Workman S, Symes A, et al. Altered microbiota, impaired quality of life, malabsorption, infection, and inflammation in CVID patients with diarrhoea. Front Immunol. 2020;11:1654. [CrossRef]
Ahmed A, King W, Sharma A. Gastrointestinal manifestations of common variable immunodeficiency: a mentored review. Dig Dis Sci. 2025 Jun 2. [CrossRef]
Malamut G, Verkarre V, Suarez F, Viallard JF, Lascaux AS, Cosnes J, et al. The enteropathy associated with common variable immunodeficiency: the delineated frontiers with celiac disease. Am J Gastroenterol. 2010;105(10):2262-75. [CrossRef]
Maarschalk-Ellerbroek LJ, Oldenburg B, Mombers IM, Hoepelman AI, Brosens LA, Offerhaus GJ, et al. Outcome of screening endoscopy in common variable immunodeficiency disorder and X-linked agammaglobulinemia. Endoscopy. 2013;45(4):320-3. [CrossRef]
Pikkarainen S, Martelius T, Ristimäki A, Siitonen S, Seppänen MRJ, Färkkilä M. A high prevalence of gastrointestinal manifestations in common variable immunodeficiency. Am J Gastroenterol. 2019;114(4):648-55. [CrossRef]
van der Poorten DK, McLeod D, Ahlenstiel G, Read S, Kwok A, Santhakumar C, et al. Gastric cancer screening in common variable immunodeficiency. J Clin Immunol. 2018;38(7):768-77. [CrossRef]
Biagi F, Bianchi PI, Zilli A, Marchese A, Luinetti O, Lougaris V, et al. The significance of duodenal mucosal atrophy in patients with common variable immunodeficiency: a clinical and histopathologic study. Am J Clin Pathol. 2012;138(2):185-9. [CrossRef]
Venhoff N, Emmerich F, Neagu M, Salzer U, Koehn C, Driever S, et al. The role of HLA DQ2 and DQ8 in dissecting celiac-like disease in common variable immunodeficiency. J Clin Immunol. 2013;33(5):909-16. [CrossRef]
Chawla S, Barman P, Tyagi R, Jindal AK, Sharma S, Rawat A, et al. Autoimmune cytopenias in common variable immunodeficiency are a diagnostic and therapeutic conundrum: an update. Front Immunol. 2022;13:869466. [CrossRef]
Feuille EJ, Anooshiravani N, Sullivan KE, Fuleihan RL, Cunningham-Rundles C. Autoimmune cytopenias and associated conditions in CVID: a report from the USIDNET Registry. J Clin Immunol. 2018;38(1):28-34. [CrossRef]
Arieira C, Dias de Castro F, Moreira MJ, Cotter J. Common variable immunodeficiency-associated inflammatory enteropathy: The new era of biological therapy. GE Port J Gastroenterol. 2018;25(6):322-6. [CrossRef]
Hashash JG, Squire J, Francis FF, Binion DG, Cross RK, Farraye FA. An expert opinion/approach: Clinical presentations, diagnostic considerations, and therapeutic options for gastrointestinal manifestations of common variable immune deficiency. Am J Gastroenterol. 2022;117(11):1743-52. [CrossRef]
Agarwal S, Mayer L. Diagnosis and treatment of gastrointestinal disorders in patients with primary immunodeficiency. Clin Gastroenterol Hepatol. 2013;11(9):1050-63. [CrossRef]
Jha P, Elangovan A, Chavan AK, Cooper G, Katz J, Cominelli F, et al. The natural history of inflammatory bowel disease in adults with common variable immunodeficiency: A case series from a single US tertiary care center. Crohns Colitis 360. 2025;7(1):otae062. [CrossRef]
Rosario NA, Rosario CS, Neto HJC, Riedi CA. Oral immunoglobulin controls chronic diarrhea in common variable immunodeficiency (CVID). J Allergy Clin Immunol. 2017;139(2):AB219.
Ahmed Z, Imdad A, Connelly JA, Acra S. Autoimmune enteropathy: An updated review with special focus on stem cell transplant therapy. Dig Dis Sci. 2019;64(3):643-54. [CrossRef]
Ciccocioppo R, Russo ML, Bernardo ME, Biagi F, Catenacci L, Avanzini MA, et al. Mesenchymal stromal cell infusions as rescue therapy for corticosteroid-refractory adult autoimmune enteropathy. Mayo Clin Proc. 2012;87(9):909-14. [CrossRef]
VOLUME
,
ISSUE
Correspondence
Received
Accepted
Published
Suggested Citation
DOI
License
