Integrated Clinical and In Silico Analysis of Hypomorphic DCLRE1C Variants: Predicting Malignancy and Autoimmunity Risk










Abstract
Objective:
Hypomorphic variants of DNA cross-link repair 1C (DCLRE1C) cause a spectrum of combined immunodeficiency phenotypes that range from mild autoimmunity to life-threatening malignancy. We aimed to integrate detailed clinical phenotyping with in silico pathogenicity metrics to improve the prediction of adverse outcomes in this rare disorder.
Materials and Methods:
We compiled a descriptive cohort of 41 patients carrying one of 12 published hypomorphic DCLRE1C variants. Inferential analyses focused on the molecularly homogeneous subgroup of 25 individuals harboring three recurrent missense changes within the metallo-β-lactamase/β-CASP domains—c.194C>T [p.Thr65Ile], c.500C>T [p.Thr167Met] and c.632G>T [p.Gly211Val]. Comprehensive clinical, immunological, and laboratory data were extracted. Pathogenicity was assessed with eight in silico algorithms, ΔΔG stability modelling, and structural mapping. Logistic regression identified variables associated with malignancy and autoimmunity, and composite risk scores were generated.
Results:
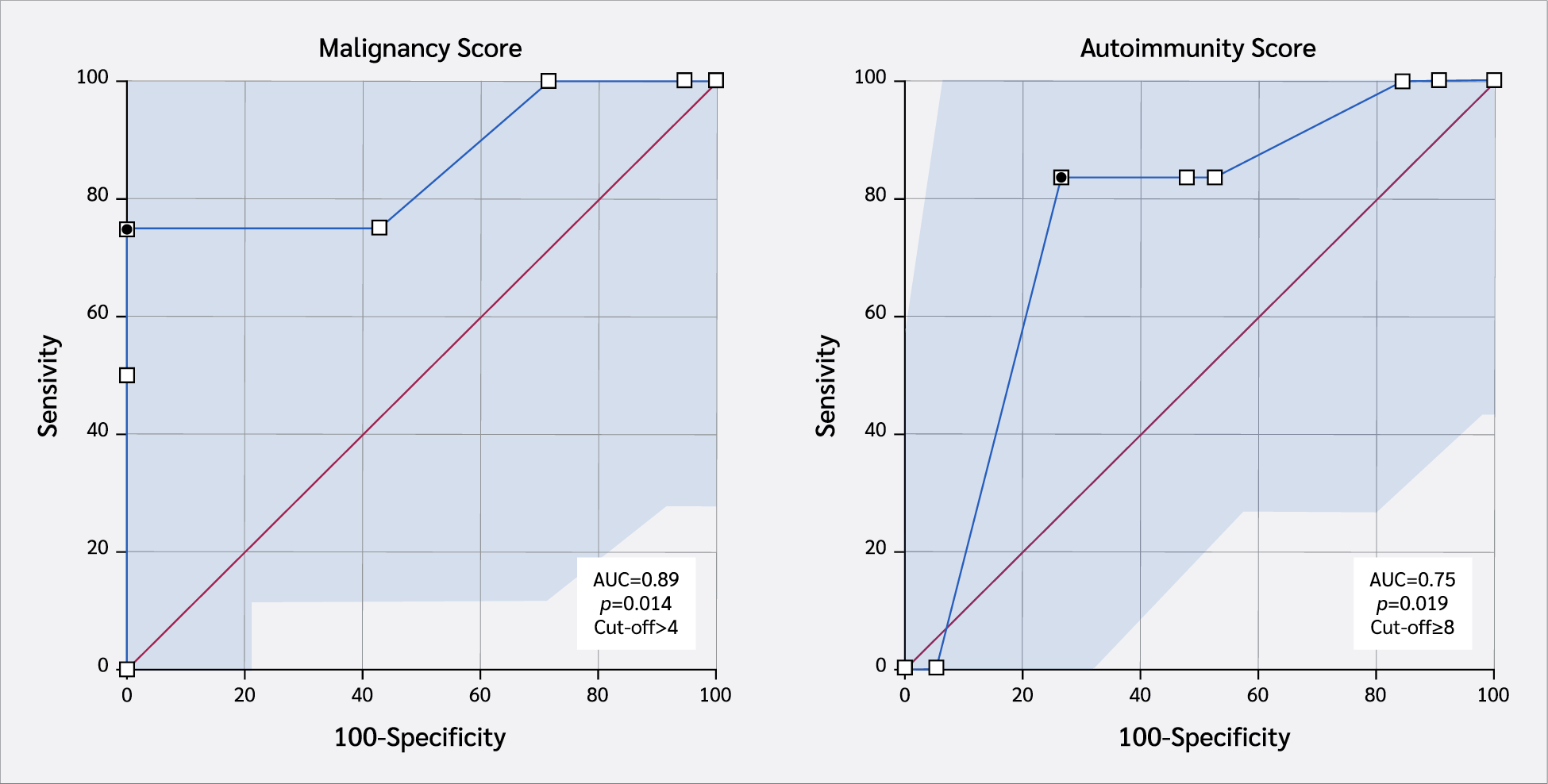
Median age at last follow-up was 9.4 years (range 0.5–27). Recurrent sinopulmonary infection (80%), chronic diarrhea (48%), and candidiasis (44%) were common across variants, while otitis media and growth retardation were confined to c-terminal changes. All three analyzed variants exceeded pathogenicity thresholds (mean Combined Annotation-Dependent Depletion [CADD] 26.7 ± 2.1) and reduced predicted protein stability (mean ΔΔG +1.8 kcal/mol). A revised malignancy risk score (lymphadenopathy, immunoglobulin G [IgG] <400 mg/dL, diarrhea, ΔΔG ≥ 1 kcal/mol, BayesDel >0.5, age > 60 months) > 4 achieved an area under the receiver operating characteristic curve (AUC) of 0.89 (95% confidence interval [CI] 0.57–1.00), with 75% sensitivity and 100% specificity. The analogous autoimmunity score (skin lesions, pulmonary infections, IgG low, ΔΔG ≥ 1, BayesDel > 0.5, CD19 low, age > 60 months) > 8 showed moderate discrimination (AUC 0.75; 95% CI, 0.54–0.97), with 83.3% sensitivity and 73.7% specificity.
Conclusion:
Integration of domain-restricted in silico metrics with core clinical parameters modestly improves malignancy risk prediction in hypomorphic DCLRE1C deficiency. Prospective, multi-center validation and functional assays are required before clinical implementation.
Keywords:
Artemis leaky, severe, combined, immunodeficiency combined, immune, deficiency DCLRE1CIntroduction
The Artemis protein, encoded by the DNA cross-link repair 1C (DCLRE1C) gene, plays a critical role in variable, diversity, and joining [V(D)J] recombination and in the repair of non-homologous end joining (NHEJ) DNA double-strand breaks (1,2). Pathogenic variants in the DCLRE1C gene severely impair T and B-cell development, resulting in the clinical phenotype of severe combined immunodeficiency (SCID) (3). In contrast, hypomorphic DCLRE1C variants allow for partial protein expression, leading to a milder phenotype of combined immunodeficiency (CID) (4).
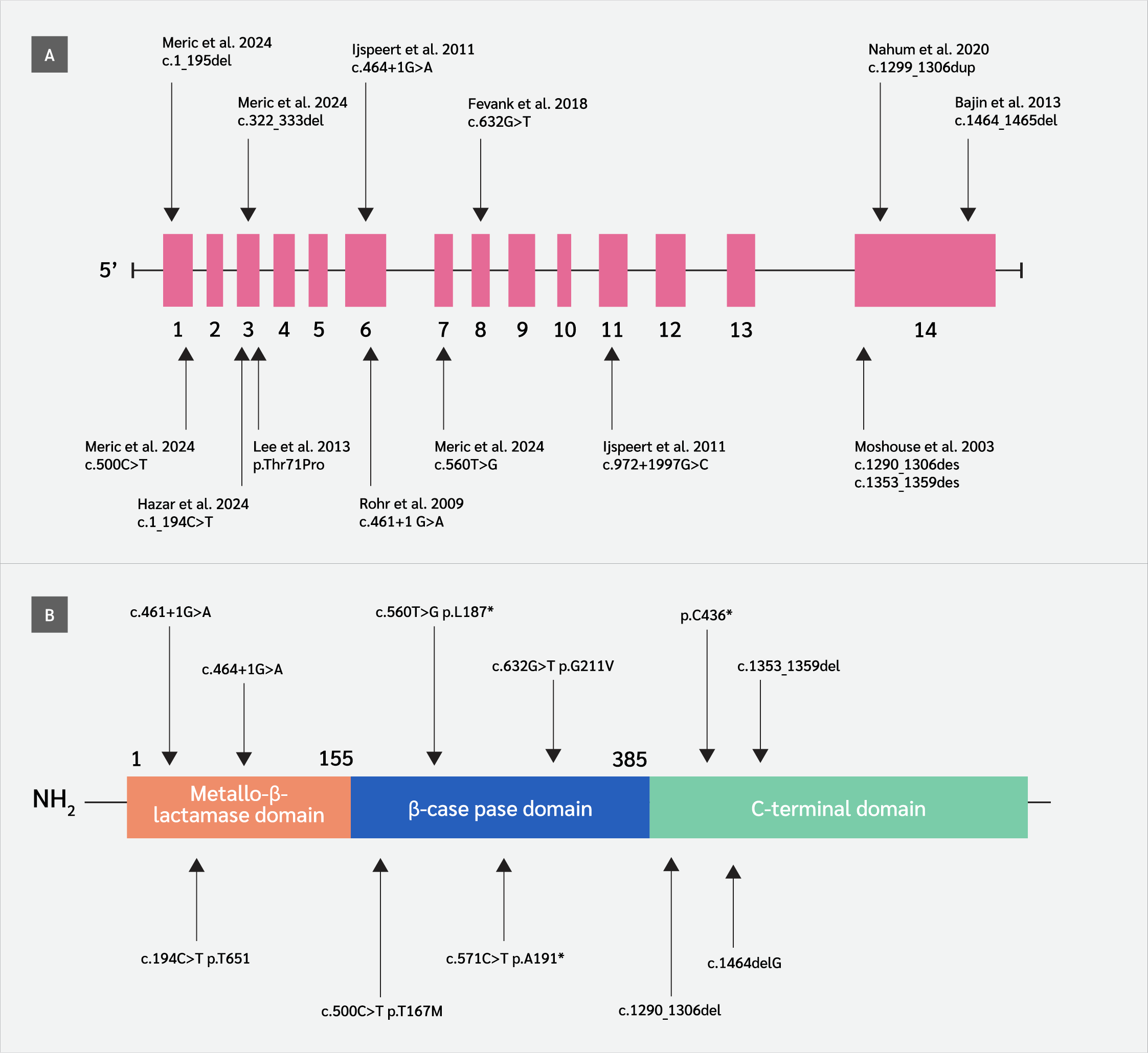
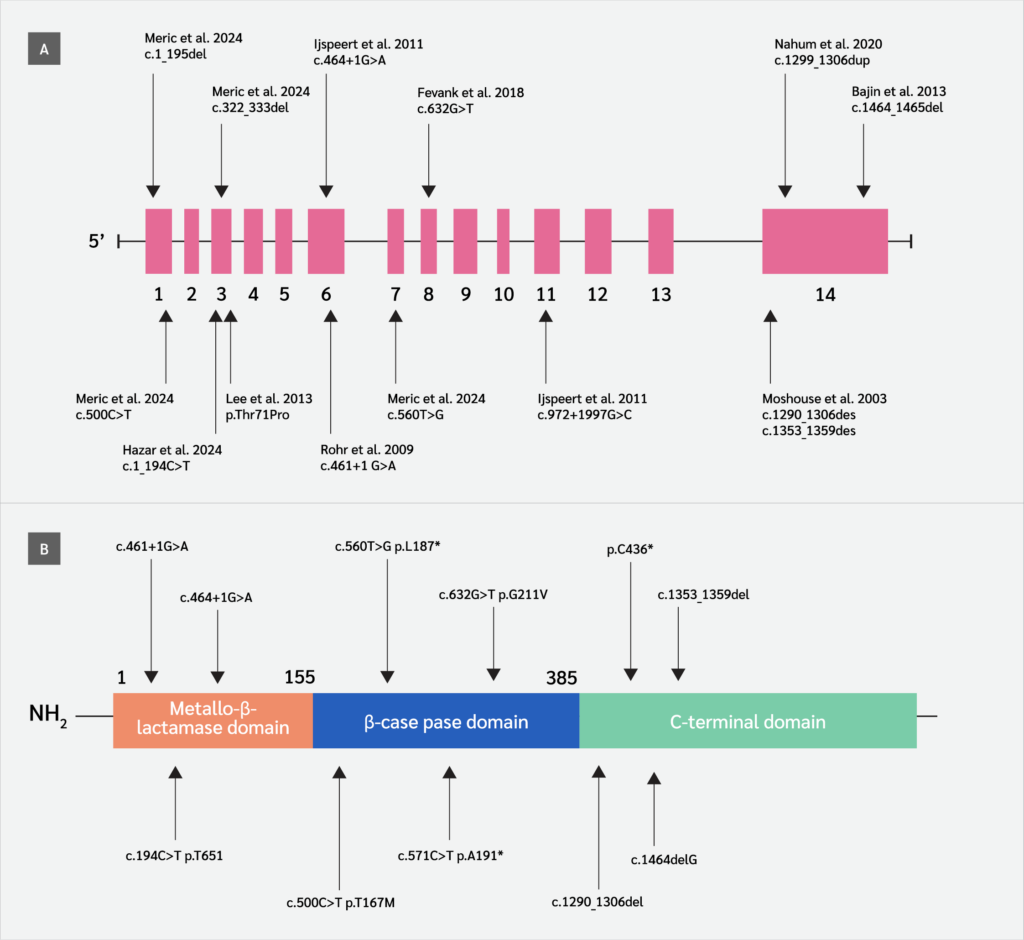
Patients with hypomorphic DCLRE1C variants, first identified in 2015 (5) may present at variable ages with diverse clinical manifestations (infections, granulomatous lesions, autoimmune disorders, malignancy) and laboratory findings. To date, only 16 hypomorphic variants in the DCLRE1C gene have been reported in 41 patients, resulting in a CID phenotype (Figure 1) (5-13). In addition to CID, these patients often develop malignancies and autoimmune diseases later in life (13), and predicting these conditions remains highly challenging. No large case series has been published regarding patients affected by the hypomorphic DCLRE1C gene variant, and the cases in the literature consist of studies with a small number of cases. Despite their clinical relevance, functional impacts of hypomorphic variants remain unclear. There is limited research investigating the correlation between hypomorphic DCLRE1C variants and clinical or laboratory parameters. Prior research has mainly focused on functional validations, creating a knowledge gap in the application of in silico tools for pathogenicity prediction in this context (14,15). Recently, in silico tools have emerged as promising approaches for predicting the pathogenicity of variants detected in patients (16-19).
Inborn errors of immunity (IEI) are rare diseases caused by mutations in over 550 genes (20,21), many of which are still classified as variants of uncertain significance (VUS), posing challenges for clinicians. However, pathogenicity analyses of variants identified as VUS through in silico analyses may lead to an increase in the number of new genes responsible for IEI, providing patients with the opportunity for early treatment. In IEI patients, variants identified as VUS have begun to be demonstrated as pathogenic through in silico analyses followed by functional studies (16-19).
From this perspective, our study aimed to correlate clinical and laboratory findings with data obtained through in silico analyses of hypomorphic DCLRE1C variants described in the literature. We further suggest that this approach may be applicable to similar analyses in other genes associated with IEI, especially when functional assays are unavailable.
Materials and Methods
Study Design
The study employed a two-tier design and was conducted at the Department of Paediatric Immunology and Allergy, Necmettin Erbakan University Faculty of Medicine.
We assembled a descriptive cohort of 41 patients (23 from the literature, 18 from our center) who harbor one of 12 hypomorphic DCLRE1C variants that have been reported to date (5-13). For genotype-phenotype comparisons, data from all available patients were included to provide a comprehensive descriptive overview. However, for laboratory-based in silico analyses, cases from the β-CASP domain were excluded due to substantial missing information. To maintain analytic homogeneity, these analyses were therefore restricted to 25 patients carrying missense variants in the metallo-β-lactamase (MBL)/β-CASP domains (c.194C>T [p.Thr65Ile], c.500C>T [p.Thr167Met], and c.632G>T [p.Gly211Val]). Among these 25 patients, 18 originated from our institutional cohort and seven from the literature.
C-terminal variants were excluded from in silico analyses because they predominantly consist of large deletions, insertions, or duplications, which cannot be reliably evaluated using tools. These cases were nevertheless retained for descriptive clinical analyses.
In silico Analysis of Commonly Observed Hypomorphic DCLRE1C Variants
A comprehensive literature review was conducted to identify pathogenic and potentially pathogenic variants associated with the Artemis protein, as shown in Figure 1. To assess the pathogenicity of these variants, we calculated multiple in silico prediction scores, including BayesDel addAF, BayesDel noAF, Combined Annotation-Dependent Depletion (CADD), Eigen, Eigen-PC, Functional Analysis through Hidden Markov Models (FATHMM)- Multiple Kernel Learning (MKL), FATHMM- eXtended Features (XF), and Deleterious Annotation of Genetic Variants using Neural Networks (DANN) (22). These scores provide insights into the functional impact of the variants based on different computational models that integrate sequence conservation, protein structure, and functional annotations.
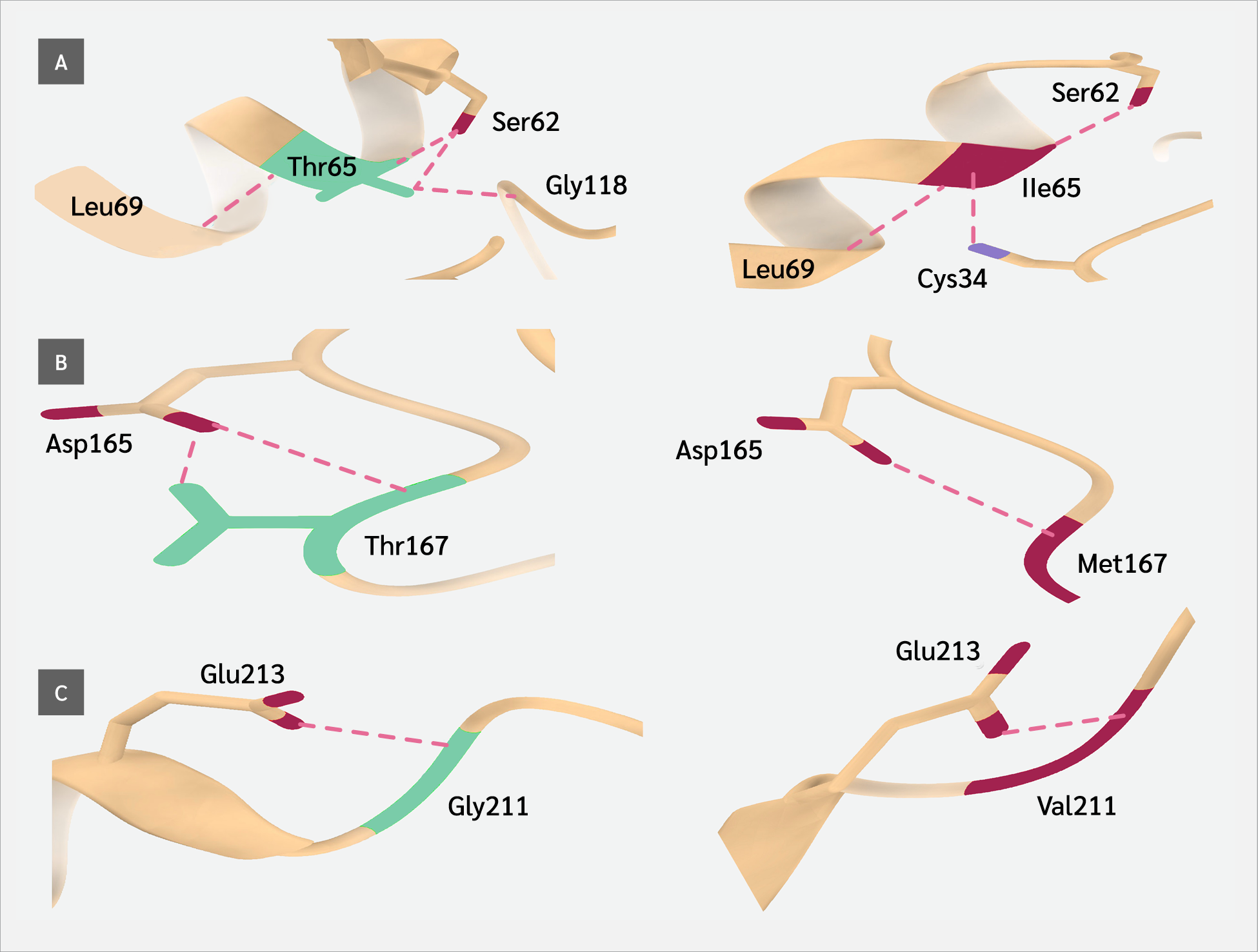
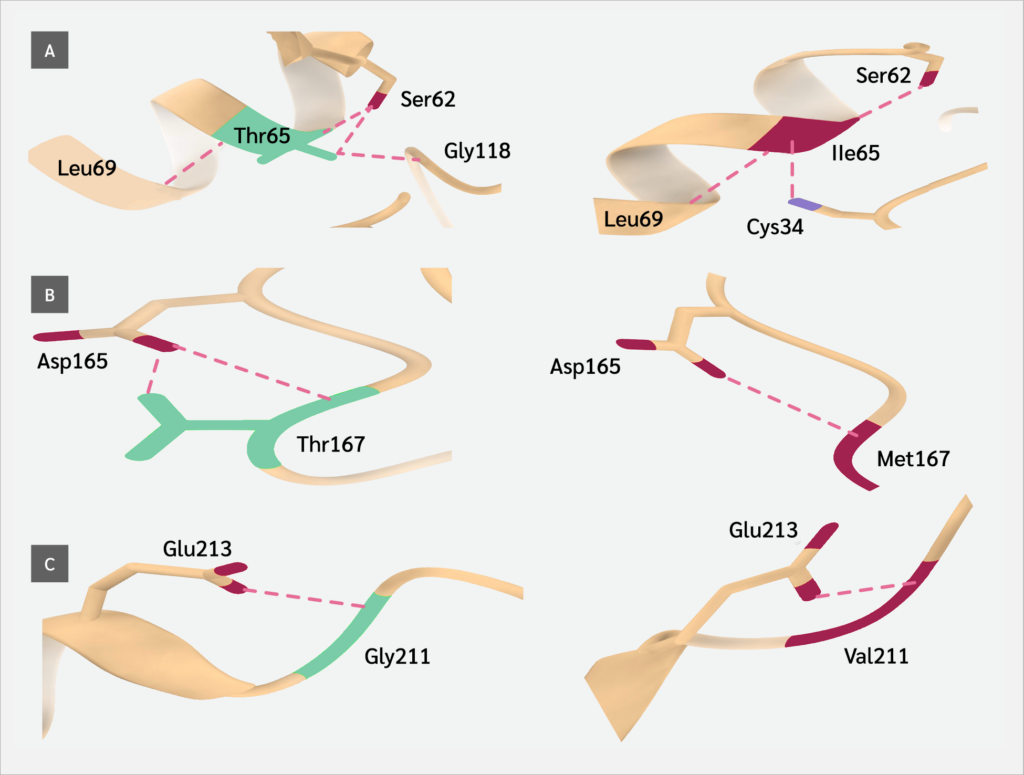
The three-dimensional (3D) structure of the Artemis protein was retrieved from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB ID: 6WO0; https://www.rcsb.org). To evaluate the structural consequences of the identified variants, we utilized PremPS (Li Lab, Soochow University, China; http://lilab.jysw.suda.edu.cn/research/PremPS/), a tool designed to predict the impact of amino acid substitutions on protein stability (23). This analysis enabled us to determine the delta-delta G (ΔΔG) (kcal/mol) values, representing the predicted change in protein stability induced by each variant. A positive ΔΔG value indicates destabilization, whereas a negative value suggests stabilization of the protein structure.
Wild-type and mutant structures were compared using UCSF ChimeraX (version 1.10.1; Resource for Biocomputing, Visualization, and Informatics, University of California, San Francisco, USA) (25). In addition to structural analyses, the evolutionary conservation of the affected residues was evaluated. Homologous protein sequences across multiple species were retrieved from the Ensembl database (release 115; European Bioinformatics Institute, Hinxton, UK), and multiple sequence alignment (MSA) was performed using the ClustalΩ algorithm (version 2024; EMBL–EBI, Hinxton, Cambridge, UK) within the JalView platform (version 2.11.5.0; The Barton Group, University of Dundee, UK) (26). Conservation scores were analyzed to determine whether the substituted residues are located within highly conserved regions, as alterations in such positions are more likely to be functionally deleterious.
Statistical Analysis
Categorical variables, including the presence of autoimmunity and malignancy, were compared across variant domain groups using the chi-square test of independence. Continuous variables were analyzed with the Mann-Whitney U test. Demographic data are presented as mean ± standard deviation (SD). All statistical analyses were performed using Python software (version 3.11; Python Software Foundation, Wilmington, DE, USA) and GraphPad Prism (version 6.0; GraphPad Software, San Diego, CA, USA). A two-sided p value <0.05 was considered statistically significant.
A composite risk scoring system was developed to predict the likelihood of malignancy and autoimmunity based on clinical, immunological, and in silico parameters. Variable selection was informed by univariate association, biological plausibility, and multicollinearity assessment. Each parameter was assigned a weighted score reflecting its relative contribution (Table 1). The total score ranges were optimized to 0–10 for malignancy and 0–12 for autoimmunity.

Predictive performance was evaluated using receiver operating characteristic (ROC) curve analysis. The area under the ROC curve (AUC) was calculated, and 95% confidence intervals (CIs) were estimated using the method of DeLong et al. (24). To assess overfitting and provide optimism-corrected performance estimates, bias-corrected and accelerated (BCa) bootstrap CIs were computed with 1000 replications, using a fixed random seed (seed=978) to ensure reproducibility. Optimal cut-off values were determined using the Youden index. Sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and odds ratios (ORs) with 95% CIs were calculated using 2×2 contingency tables and Fisher’s exact test where appropriate. For all estimated effect sizes, including odds ratios, 95% CIs were computed using exact or asymptotic methods as appropriate and are reported alongside p-values to reflect the precision of the estimates.
In this exploratory cohort, the predictive performance of the risk scores was evaluated in the context of a small sample size and clinical heterogeneity. Given the exploratory nature of this study and the rarity of hypomorphic DCLRE1C variants, AUC values exceeding 0.60 were considered to reflect potential discriminative ability in the context of preliminary risk stratification, with statistical significance (p<0.05 vs. AUC=0.5) and internal validation via bootstrapping serving as key supportive criteria.
Results
Genomics, Clinical, and Laboratory Characteristics of Hypomorphic DCLRE1C Variants
In the initial phase of the study, a total of 41 patients with hypomorphic DCLRE1C variants were identified through literature review and institutional records. Domain-based distribution and genotype associations were summarized as follows: Among them, 23 had mutations in the MBL domain, 15 in the C-terminal domain, and 3 in the β-CASP domain. Genotypic analysis revealed that 34 (82.9%) patients were homozygous, and 7 (17.1%) were compound heterozygous patients. No statistically significant differences were observed in the frequency of autoimmunity across variant domains (p=0.470). Similarly, malignancy rates did not differ significantly among the domain groups (p=0.297). In pairwise comparisons between genotypes, the prevalence of autoimmunity was similar between homozygous and compound heterozygous patients as was the occurrence of malignancy (p>0.05). These findings suggest that neither the structural localization of the DCLRE1C variant nor genotype (homozygous vs compound heterozygous) significantly influences the risk of autoimmunity or malignancy in this cohort.
Among the clinical features examined, the frequency of pulmonary infections was significantly higher in patients with MBL domain variants (63.2%) compared to those with C-terminal domain variants (p=0.032), whereas the difference between MBL and β-CASP domains was not statistically significant (p=0.236), nor was the difference between β-CASP and C-terminal domains (p=0.502). Several other clinical features showed statistically significant differences across domain groups, including diarrhea (p=0.028), otitis (p=0.038), fever (p<0.001), growth retardation (p=0.007), malignancy (p=0.048), and mortality status (p=0.006); however, skin lesions did not differ significantly. Notably, both otitis and growth retardation were observed exclusively in the C-terminal domain group and were absent in the MBL and β-CASP groups. When clinical findings were evaluated by genotype, candidiasis was significantly more common among patients with compound heterozygous variants (3/7; 42.9%) than in those with homozygous mutations (1/34; 2.9%) (p=0.001).
Statistically significant differences were found across domain groups for natural killer (NK) cells, CD25 activation, and naïve B-cell levels. Natural killer cells were low in 3/9 C-terminal cases (33%), but normal in 0/21 MBL and 6/7 β-CASP cases (p=0.025) (12). CD25 activation in T cells was markedly reduced in both C-terminal (88.9%) and MBL (90.5%) domain groups. Although these two domains showed comparable suppression rates (p=0.894), the overall distribution reached statistical significance (p=0.030), primarily due to the single β-CASP patient exhibiting normal CD25 activation. Naïve B-cell levels differed significantly across protein domains (χ²=7.843, p=0.005). Only patients with either decreased or normal naïve B-cell levels were included in the analysis due to the absence of elevated cases. The majority of patients with MBL domain variants exhibited decreased naïve B-cell counts (94.4%), whereas most patients with C-terminal domain variants showed normal counts (66.7%).
In Silico Analysis Results
Combined Annotation–Dependent Depletion scores were available for a subset of variants located within the MBL and β-CASP domains. All variants in both domains exceeded the commonly accepted pathogenicity threshold (CADD ≥ 20) (15), indicating a 100% predicted deleterious rate for these domains. No CADD scores were available for variants in the C-terminal domain, primarily due to the presence of large deletions and complex mutations not compatible with current CADD scoring models. According to BayesDel (addAF) scores, 50% of the variants in the MBL domain and 33.3% of those in the β-CASP domain were predicted to be deleterious (threshold> 0.5). Based on Eigen scores, all evaluated variants in the MBL and β-CASP domains exhibited values greater than zero, which is indicative of potential deleteriousness. This corresponds to a 100% predicted pathogenicity rate in both groups. No Eigen scores were available for variants located in the C-terminal domain, primarily due to structural or complex variant types that are beyond the scope of this prediction tool. All evaluated variants within the MBL and β-CASP domains showed DANN scores above 0.9, indicating a predicted deleteriousness rate of 100% in both domains. Variants from the C-terminal domain could not be assessed due to unavailable scores. According to the FATHMM-MKL model, all variants in the MBL domain and 66.7% of those in the β-CASP domain were predicted to be deleterious (threshold >0.5). Using the FATHMM-XF model, deleterious predictions were observed in 50.0% and 66.7% of variants within the MBL and β-CASP domains, respectively. Based on protein stability predictions (ΔΔG ≥1.0), 50.0% of the evaluated variants in the MBL domain were classified as destabilizing, whereas none of the variants in the β-CASP domain met this threshold.
Association Between in Silico Deleteriousness Scores and Clinical Features
To explore the association between in silico pathogenicity metrics and clinical phenotypes, we developed two separate minimal predictive scoring systems tailored for malignancy and autoimmunity. Each scoring system combined selected clinical and immunological features with in silico deleterious parameters.
For malignancy risk stratification, the scoring model included lymphadenopathy, diarrhea, skin lesions, low IgG levels, ΔΔG ≥1.0, BayesDel addAF >0.5, and age >60 months. Each variable contributed one or two points depending on presumed clinical relevance, with a total possible score of 11. Receiver operating characteristic analysis in our cohort (n=25) revealed an AUC of 0.89. At an optimal cut-off of >4 points, determined by Youden’s index, the model achieved 75% sensitivity and 100% specificity, with a PPV of 100%, NPV of 95.5%, and an odds ratio of 100 (Table 2). These findings support the utility of this score in identifying patients at elevated risk of malignancy while minimizing false positives.

For autoimmunity prediction, a separate composite score was optimized using ΔΔG ≥1.0, BayesDel addAF >0.5, low IgG, low CD19 expression, and the presence of skin lesions and pulmonary infections. At a cut-off of >8 points, the model demonstrated 83.3% sensitivity and 73.7% specificity, with a PPV of 50%, NPV of 93.14%, odds ratio of 14, and an AUC of 0.75 (Table 2). Although the discriminatory power was limited, this score may serve as a high-sensitivity screening tool for identifying patients at risk of autoimmune manifestations.
Both scoring systems integrate clinical and computational evidence, offering a practical framework for early risk stratification in patients harboring hypomorphic DCLRE1C mutations.
Discussion
This study represents one of the most comprehensive evaluations of patients with hypomorphic DCLRE1C mutations under study to date, integrating clinical, immunological, and bioinformatic data. While previous reports have described these variants in small patient cohorts, our findings further highlight the significant phenotypic heterogeneity associated with hypomorphic mutations in the DCLRE1C gene. Affected individuals presented with a wide clinical spectrum, ranging from recurrent infections and autoimmune manifestations to malignancies, whereas some patients exhibited only mild features of immune deficiency. This variability suggests that DCLRE1C mutations alone may not fully account for disease expression, and that additional genetic, environmental, or epigenetic modifiers likely play a role. Furthermore, the increasing application of in silico tools provides valuable insights into the functional consequences of these variants, supporting their utility in clinical diagnostics and variant interpretation.
The pronounced phenotypic variability observed among patients with similar DCLRE1C variants suggests that additional modifying factors likely contribute to disease expression. These may include genetic background, epigenetic regulation, or environmental influences such as infectious exposures and treatment differences. Similar modifying effects have been described in other inborn errors of immunity, where host genetics, epigenetic programming, and external triggers modulate clinical severity beyond the primary causal mutation (20,21). A deeper exploration of these modifiers—through multi-omic approaches and longitudinal environmental monitoring—will be essential to fully understand disease heterogeneity in hypomorphic DCLRE1C deficiency.
Our analysis revealed no statistically significant association between the localization of these variants within functional Artemis domains and the occurrence of autoimmunity or malignancy. Similarly, genotype distribution (homozygous vs. compound heterozygous) did not predict clinical severity with respect to these outcomes. These results align with previous reports documenting phenotypic variability among patients with structurally similar mutations (5,9,13). Moshous et al. (3,4) also noted that patients with identical or similar mutations may exhibit widely differing clinical courses, suggesting the involvement of additional modifying genetic or environmental factors. Hence, while domain-based classification offers structural insights, it appears insufficient for predicting disease progression or associated complications. These findings underscore the need for integrative approaches beyond sequence localization—incorporating immune profiling, environmental exposures, and multi-omic analyses—to better predict clinical outcomes in Artemis deficiency.
Previous studies have highlighted the susceptibility to recurrent respiratory and fungal infections in patients with hypomorphic variants, though the correlation between clinical features and mutation localization has remained largely unexplored. For instance, Volk et al. (5) and Hazar et al. (13) reported recurrent sinopulmonary infections and candidiasis as common manifestations, particularly in early-onset cases. In our study, a statistically significant association was observed between C-terminal domain mutations and a higher frequency of pulmonary infections. This may reflect the functional role of the C-terminal region in facilitating nuclear localization signals and protein stability, suggesting that alterations in this domain impair mucosal immune defense more severely than mutations in catalytic regions. In addition to these findings, other clinical manifestations such as diarrhea, otitis, fever, growth retardation, malignancy, and survival status also differed significantly across domain groups. Otitis and growth retardation, notably, were observed only in patients with C-terminal domain mutations, further supporting domain-specific clinical vulnerability. Moreover, candidiasis was significantly more frequent in patients with compound heterozygous variants compared to homozygous individuals (42.9% vs. 2.9%, p=0.001), indicating that allelic diversity may exacerbate susceptibility independently of domain localization. Although candidiasis has been described in Artemis-deficient individuals (10), our data indicate that allelic diversity may exacerbate susceptibility, potentially through additive or synergistic loss of function (9). This observation warrants further investigation in functional assays and may have implications for early clinical monitoring in compound heterozygous patients.
Immunological analyses revealed domain-specific alterations in lymphocyte subsets and immune function. Notably, NK cells were reduced exclusively in patients with C-terminal domain mutations (33.3%; 3/9), while they remained normal in patients harboring variants in the MBL or β-CASP domains (p=0.028). Although no major NK cell abnormalities have been consistently reported in patients with the hypomorphic DCLRE1C variant, this finding may reflect subtle defects in hematopoietic regulation or lineage development in the context of structurally disruptive variants. Previous studies, such as Moshous et al. (4), have reported variable T- and B-cell abnormalities, but NK cell dysregulation has not been systematically analyzed. Similarly, CD25 activation, which reflects T cell proliferative capacity, decreased in most patients across domains. This aligns with findings by Rohr et al. (6) and Ijspeert et al. (7), who demonstrated impaired T cell responses in patients with splicing or deletional DCLRE1C variants. These data support the notion that even hypomorphic variants can lead to functional T cell compromise despite partial V(D)J recombination activity. These immunophenotypic differences across domains may have important clinical implications. The selective reduction of NK cells in patients with C-terminal variants could suggest a role for this region in hematopoietic lineage stability or NK cell development, potentially contributing to increased susceptibility to viral infections or malignancy. Similarly, the consistent decrease in CD25 activation across MBL and C-terminal variants reflects impaired T-cell proliferative capacity, which may compromise immune surveillance and predispose to autoimmunity or lymphoproliferation. The depletion of naïve B cells in MBL variants highlights the catalytic domain’s importance for B-cell maturation and bone marrow output, consistent with impaired V(D)J recombination activity. Taken together, these findings support the concept that distinct structural regions of Artemis differentially influence lymphocyte subsets, providing mechanistic clues to the heterogeneous clinical outcomes observed in hypomorphic DCLRE1C deficiency.
The most striking immunological finding was the depletion of naïve B-cells. All patients with MBL domain mutations exhibited reduced naïve B-cells (100%), whereas those with C-terminal or β-CASP variants displayed mixed patterns. Since naïve B-cells are a critical marker of bone marrow output and immune reconstitution, this domain-specific depletion suggests that catalytic domain integrity may be particularly essential for B cell development. These results expand upon prior observations by Volk et al. (5) and highlight the diagnostic value of detailed immunophenotyping in Artemis-deficient cohorts.
Our in silico analysis demonstrated consistently high pathogenicity scores for missense variants located in the MBL and β-CASP domains, with all assessed mutations surpassing deleteriousness thresholds for CADD, DANN, and Eigen scores. This is consistent with the catalytic importance of these regions, where even subtle amino acid substitutions may severely impair Artemis function. In contrast, variants located in the C-terminal domain were not amenable to assessment using most computational models, largely due to their complex nature (e.g., large deletions or splice-disrupting mutations). This highlights a technical limitation of current in silico tools, which remain optimized for simple missense changes rather than structural alterations or non-coding variants (14,15).
Protein stability analysis using ΔΔG values (PremPS) further reinforced these findings: half of the variants in the MBL domain were predicted to destabilize the protein structure (ΔΔG ≥1.0), whereas no such destabilization was observed in β-CASP domain variants. This discrepancy underscores that not all high-scoring in silico predictors necessarily translate into structural damage, reaffirming the importance of integrating multiple tools and domains of evidence. Similar approaches have recently been validated in other primary immunodeficiencies, such as ISG15 and ZBTB24-related ICF (immunodeficiency, centromeric instability, and facial anomalies) syndromes (16,18), supporting the broader utility of in silico-guided pathogenicity frameworks in rare immune disorders.
The incorporation of in silico predictors into the model is particularly noteworthy, as these tools are increasingly being used to reclassify VUS in primary immunodeficiencies (16,17). Building on this trend, our study introduces a novel composite scoring system tailored to Artemis deficiency. To our knowledge, no comparable composite scoring system has been proposed in the context. Existing studies tend to describe qualitative associations or focus solely on molecular characterization (5,13), without integrating predictive algorithms into patient management strategies.
To improve clinical risk stratification in hypomorphic DCLRE1C variant carriers, we developed a minimal predictive score incorporating both clinical and in silico variables. Although preliminary, the model demonstrated modest discriminatory power for malignancy and autoimmunity, with particularly high sensitivity for malignancy at a threshold of >4 points. Although specificity was limited, the high sensitivity suggests that the score may be useful as a screening tool for early risk stratification. In interpreting these results, it is important to note that variable weightings were intentionally guided by presumed clinical relevance rather than purely statistical optimization. Features such as lymphadenopathy and hypogammaglobulinemia were prioritized, given their established links with immunodeficiency-associated malignancy, whereas in silico parameters were assigned lower weights reflecting their exploratory role. Likewise, the malignancy cut-off of >4 points was chosen not only by statistical optimization (Youden index) but also because it represents the co-occurrence of several clinically meaningful abnormalities. This dual rationale strengthens the interpretability of the proposed score despite its exploratory nature.
Study Limitations
Although this study offers a detailed picture of the clinical impact of hypomorphic DCLRE1C variants, several constraints must be acknowledged. First, the sample size is relatively small, and a substantial proportion of cases were extracted from the literature, introducing potential publication bias and inter-center diagnostic heterogeneity. This limitation must also be considered in light of the exceptional rarity of hypomorphic DCLRE1C deficiency: to date, only 16 hypomorphic variants have been reported in 41 patients worldwide. Consequently, while our findings provide a useful preliminary framework, the limited cohort size reduces statistical power and precludes external validation. Future prospective, multi-center collaborations will therefore be indispensable to confirm and refine the predictive models.
Second, the inclusion of both institutional and literature-derived cases may still introduce heterogeneity in clinical assessments. However, to mitigate this effect, we applied strict inclusion criteria, excluded literature cases with substantial missing data, and restricted inferential analyses to a genetically homogeneous subgroup. Despite these steps, residual heterogeneity remains a potential source of bias.
Third, the in silico pathogenicity and protein stability predictions incorporated into our analysis have not yet been validated by functional assays such as DNA repair activity or V(D)J recombination studies. Although prior reports have demonstrated functional impairment in hypomorphic DCLRE1C variants, our predictive models remain inferential until corroborated by direct experimental validation.
In addition, this study may be subject to selection bias, as literature-derived cases were included based on available data quality, which may not fully represent the entire spectrum of hypomorphic DCLRE1C deficiency. Missing data for certain clinical and immunological parameters further limited statistical power and comparability across groups. Finally, the retrospective design inherently introduces heterogeneity in diagnostic assessments and follow-up practices across centers, which should be considered when interpreting the results.
Conclusion
Hypomorphic DCLRE1C variants exhibit broad clinical variability, and current structural or genotypic classifications alone appear insufficient for precise risk stratification. Integrating domain-restricted in silico metrics with clinical parameters modestly improves the prediction of malignancy risk and may also provide insight into overall disease severity. Our findings suggest that combining computational predictions with clinical parameters can facilitate the earlier identification of high-risk patients and guide therapeutic decision-making -particularly in resource-limited settings where functional assays are unavailable. Nonetheless, prospective, multi-center validation and functional studies are essential before these approaches can be implemented in clinical practice.
Ethical Approval
The study was approved by the Necmettin Erbakan University Clinical Research Ethics Committee on May 23, 2025, with the decision number 5789.
Informed Consent
N.A.
Peer-review
Externally peer-reviewed
Author Contributions
Concept – S.K., M.A.K., İ.R.; Design – S.K., M.A.K., A.Ş.; Supervision – İ.R., S.Ke., Ş.N.G.; Data Collection and/or Processing – S.K., M.A.K., A.Ş., T.D., Ş.N.G., S.Ke.; Analysis and/or Interpretation – S.K., M.A.K., A.Ş., T.D.; Literature Review – S.K., M.A.K., T.D.; Writer – S.K., M.A.K., A.Ş., T.D.; Critical Reviews – M.A.K., Ş.N.G., S.Ke., İ.R.
Conflict of Interest
The author declares no conflict of interest.
Financial Disclosure
The authors declared that this study has received no financial support.
Acknowledgment
The authors have no acknowledgments to declare.
Scientific Presentation:
The preliminary data of this study were presented as an abstract at the 11th Clinical Immunology Congress, held in Antalya, Türkiye, on April 9–12, 2025, where the study received the third prize for oral presentation.
References
de Villartay, J. P. Congenital defects in V(D)J recombination. Br Med Bull. 2015;114(1):157-67. [CrossRef]
Buckley RH, Schiff RI, Schiff SE, Markert ML, Williams LW, Harville TO, et al. Human severe combined immunodeficiency: genetic, phenotypic, and functional diversity in one hundred eight infants. J Pediatr. 1997;130(3):378-87. [CrossRef]
Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105(2):177-86. [CrossRef]
Moshous D, Pannetier C, Chasseval Rd Rd, Deist Fl Fl, Cavazzana-Calvo M, Romana S, et al. Partial T and B lymphocyte immunodeficiency and predisposition to lymphoma in patients with hypomorphic mutations in Artemis. J Clin Invest. 2003;111(3):381-7. [CrossRef]
Volk T, Pannicke U, Reisli I, Bulashevska A, Ritter J, Björkman A, et al. DCLRE1C (ARTEMIS) mutations causing phenotypes ranging from atypical severe combined immunodeficiency to mere antibody deficiency. Hum Mol Genet. 2015;24(25):7361-72. [CrossRef]
Rohr J, Pannicke U, Döring M, Schmitt-Graeff A, Wiech E, Busch A, et al. Chronic inflammatory bowel disease as key manifestation of atypical ARTEMIS deficiency. J Clin Immunol. 2010;30(2):314-20. [CrossRef]
Ijspeert H, Lankester AC, van den Berg JM, Wiegant W, van Zelm MC, Weemaes CM, et al. Artemis splice defects cause atypical SCID and can be restored in vitro by an antisense oligonucleotide. Genes Immun. 2011;12(6):434-44. [CrossRef]
Bajin İY, Ayvaz DÇ, Ünal S, Özgür TT, Çetin M, Gümrük F, et al. Atypical combined immunodeficiency due to Artemis defect: a case presenting as hyperimmunoglobulin M syndrome and with LGLL. Mol Immunol. 2013;56(4):354-7. [CrossRef]
Lee PP, Woodbine L, Gilmour KC, Bibi S, Cale CM, Amrolia PJ, et al. The many faces of Artemis-deficient combined immunodeficiency - Two patients with DCLRE1C mutations and a systematic literature review of genotype-phenotype correlation. Clin Immunol. 2013;149(3):464-74. [CrossRef]
Fevang B, Fagerli UM, Sorte H, Aarset H, Hov H, Langmyr M, et al. Runaway train: a leaky radiosensitive SCID with skin lesions and multiple lymphomas. Case Reports Immunol. 2018:2053716. [CrossRef]
Nahum A, Somech R, Shubinsky G, Levy J, Broides A. Unusual phenotype in patients with a hypomorphic mutation in the DCLRE1C gene: IgG hypergammaglobulinemia with IgA and IgE deficiency. Clin Immunol. 2020;213:108366. [CrossRef]
Meric Z, Gemici Karaaslan B, Yalcin Gungoren E, Bektas Hortoglu M, Cavas T, Aydemir S, et al. Artemis deficiency: A large cohort including a novel variant with increased radiosensitivity. Pediatr Allergy Immunol. 2024;35(6):e14171. [CrossRef]
Hazar E, Karaselek MA, Kapakli H, Dogar O, Kuccukturk S, Uygun V, et al. Variable clinical presentation of hypomorphic DCLRE1C deficiency from childhood to adulthood. Pediatr Allergy Immunol. 2024;35(10):e14260. [CrossRef]
Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB; IARC Unclassified Genetic Variants Working Group. In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat. 2008;29(11):1327-36. [CrossRef]
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310-5. [CrossRef]
Napoleao SMDS, Salgado RC, Ferreira JFS, de Barros Dorna M, de Moura TCL, França TT, et al. First Brazilian case report of unrelated patients with identical ISG15 mutation. J Clin Immunol. 2024;45(1):21. [CrossRef]
Vorsteveld EE, Van der Made CI, Smeekens SP, Schuurs-Hoeijmakers JH, Astuti G, Diepstra H, et al. Clinical exome sequencing data from patients with inborn errors of immunity: Cohort level diagnostic yield and the benefit of systematic reanalysis. Clin Immunol. 2024;268:110375. [CrossRef]
Duran T, Karaselek MA, Kuccukturk S, Gul Y, Sahin A, Guner SN, et al. Investigation of transcription factor and cytokine gene expression levels in helper T cell subsets among Turkish patients diagnosed with ICF2 (novel ZBTB24 gene variant) and ICF3 (CDCA7 variant) syndrome. J Clin Immunol. 2024;45(1):16. [CrossRef]
Jenni R, Klaa H, Khamessi O, Chikhaoui A, Najjar D, Ghedira K, et al. Clinical and genetic spectrum of Ataxia Telangiectasia Tunisian patients: Bioinformatic analysis unveil mechanisms of ATM variants pathogenicity. Int J Biol Macromol. 2024;278(Pt 1):134444. [CrossRef]
Poli MC, Aksentijevich I, Bousfiha AA, Cunningham-Rundles C, Hambleton S, Klein C, et al. Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. J Hum Immun. 2025;1(1):e20250003. [CrossRef]
IJspeert H, Edwards ESJ, O'Hehir RE, Dalm VASH, van Zelm MC. Update on inborn errors of immunity. J Allergy Clin Immunol. 2025;155(3):740-51. [CrossRef]
Schubach M, Maass T, Nazaretyan L, Röner S, Kircher M. CADD v1.7: using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 2024;52(D1):D1143-54. [CrossRef]
Chen Y, Lu H, Zhang N, Zhu Z, Wang S, Li M. PremPS: Predicting the impact of missense mutations on protein stability. PLoS Comput Biol. 2020;16(12):e1008543. [CrossRef]
DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837-45. [CrossRef]
Meng EC, Goddard TD, Pettersen EF, Couch GS, Pearson ZJ, Morris JH, et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 2023;32(11):e4792. [CrossRef]
Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20(3):426-7. [CrossRef]
VOLUME
,
ISSUE
Correspondence
Received
Accepted
Published
Suggested Citation
DOI
License
